


Sommario
La sindrome di Down è dovuta alla presenza di una copia in eccesso del cromosoma 21: è una malattia genetica ma non ereditaria che si manifesta con frequenza costante nelle diverse aree del mondo. Benché le cause di questa alterazione non siano ancora state chiarite, la scienza ha individuato il più importante fattore di rischio, l’età materna.
La malattia è caratterizzata da un ritardo dello sviluppo fisico e mentale, un aspetto specifico di testa, viso, mani e piedi e da complicanze che interessano un ampio gruppo di sistemi e apparati (fra cui leucemia e disturbi psichiatrici).
Durante l’invecchiamento sono frequenti forme di demenza, con crisi di aggressività e collera e riduzione delle capacità motorie.
In gravidanza, alcuni segni visibili all’ecografia possono raccomandare esami come il prelievo dei villi coriali o l’amniocentesi. La diagnosi postnatale viene posta sulla base delle caratteristiche fisiche e verificata con la mappatura dei geni e dei cromosomi presenti nelle cellule.
La prospettiva di vita dei pazienti è notevolmente aumentata nel corso degli ultimi decenni, grazie alla maggiore capacità diagnostica e terapeutica delle complicanze, al livello di integrazione scolastica e inclusione sociale dei pazienti raggiunto e all’attuazione dei protocolli riabilitativi. Di pari passo sono sorti numerosi dibattiti sul tema dell’aborto.
La sindrome di Down a mosaico è una forma lieve della malattia, nella quale la persona colpita ha un numero di cellule con il difetto e un numero di cellule sane. Il quadro clinico è tanto più favorevole quanto più elevato è il numero degli elementi sani.
Sindrome di Down: che cos’è
La trisomia 21 è paradigmatica per diverse ragioni. La prima, e forse la più importante, è legata al concetto di eccesso di DNA.
Siamo abituati a sentire parlare di malattie genetiche dovute a deficit di materiale genetico, geni assenti o mutati e dunque non in grado di esercitare la loro funzione. Più complesso è immaginare una malattia così impattante sull’organismo legata alla presenza di un cromosoma supplementare.
In secondo luogo, la sindrome di Down è importante per i suoi risvolti sociali. In particolare, per gli straordinari passi avanti fatti nel miglioramento della qualità e dell’aspettativa di vita dei pazienti.
La centralità di questa patologia riguarda anche un tema spinoso come quello dell’aborto. La disponibilità di esami in grado di rivelare tempestivamente la presenza di anomalie cromosomiche gravi attribuisce una pesante responsabilità ai genitori, soprattutto alla madre, che porta in grembo il piccolo.
Esiste, poi, il tema del counselling genetico, che ha, in alcuni Paesi europei, promosso l’interruzione di gravidanza, in presenza di alterazioni cromosomiche come la trisomia 21, portando al quasi azzeramento delle nascite di bambini con questa malattia. Una scelta che ha fatto discutere perché riporta alla mente associazioni drammatiche con le teorie eugenetiche.
Infine, questa malattia mette al centro del dibattito l’opportunità femminile di concepire anche in età relativamente avanzata, resa possibile dall’aumento dell’età media e dal miglioramento degli standard di vita e di salute, ma associata ad aspetti etici non trascurabili.
La sindrome di Down e quel cromosoma in più
Questa patologia è dovuta alla presenza di un cromosoma in eccesso, che comporta lo sviluppo di manifestazioni fisiche multi-organo di diversa gravità e un ritardo mentale più o meno pronunciato.
Malgrado gli studi proseguissero da secoli, solo negli anni ’30 è stata messa in evidenza la reale causa della malattia.
Cromosomi e patrimonio genetico: la fisiologia
I cromosomi sono strutture presenti in tutte le cellule che contengono i geni. L’insieme di tutti i cromosomi rappresenta il DNA di un individuo, il suo patrimonio genetico.
In esso sono contenute le istruzioni per la produzione di tutte le proteine necessarie al corretto funzionamento dell’organismo.
Ogni gene porta scritte le informazioni per la sintesi di una proteina.
Tutti i cromosomi sono presenti in duplice copia: questo stratagemma della natura, frutto dell’evoluzione, permette di bypassare molte delle alterazioni che possono interessare il DNA. In totale i cromosomi umani sono 46.
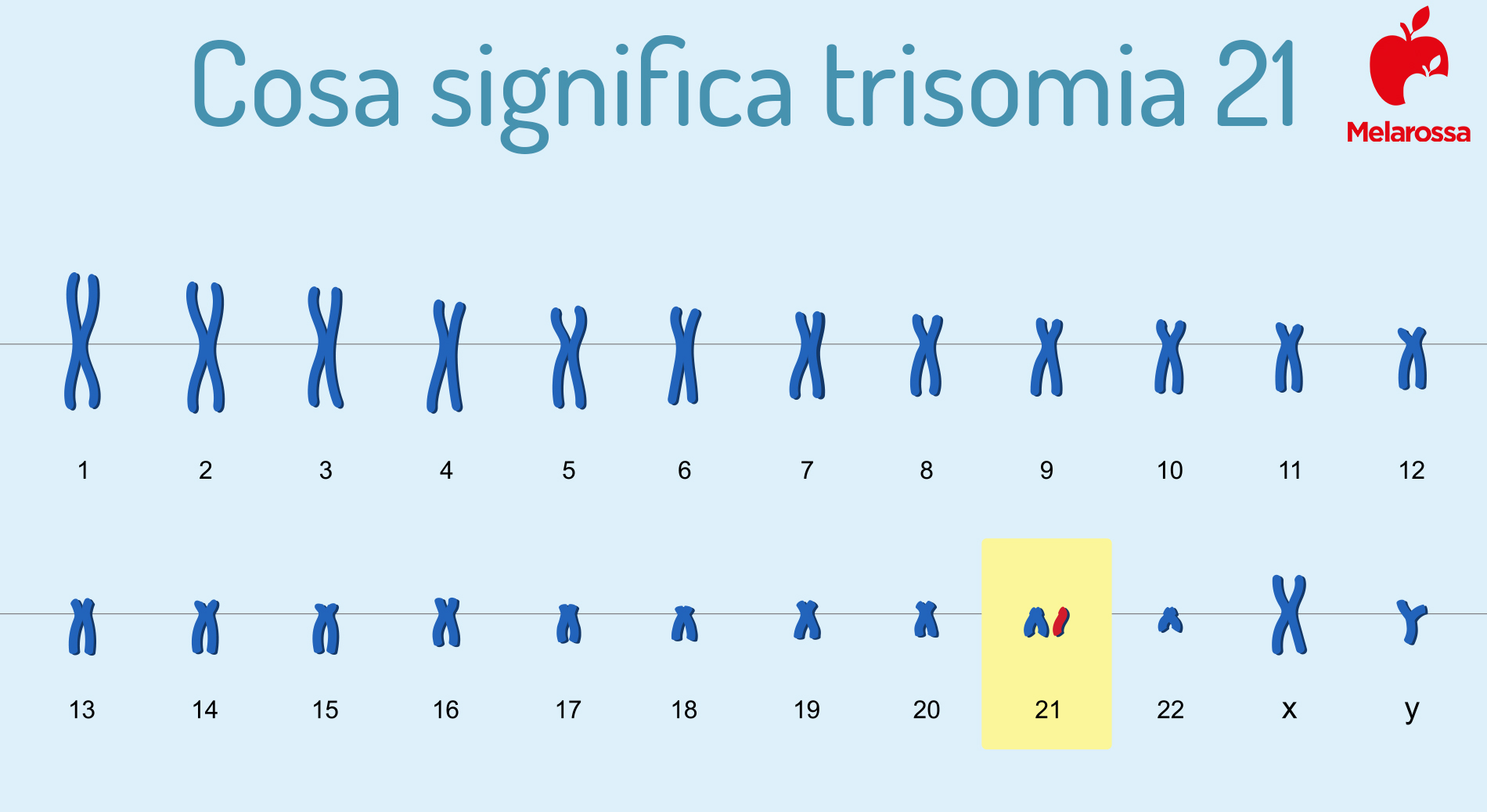
Cosa significa trisomia 21
La trisomia è la presenza di un cromosoma in triplice, anziché duplice, copia. La trisomia più comune è quella del cromosoma 21.
In generale, le trisomie portano all’interruzione spontanea della gravidanza nella quasi totalità dei casi.
Invece, nel caso del cromosoma 21, il più piccolo del patrimonio genetico umano, le sue dimensioni rendono questa grave alterazione in alcuni casi compatibile con la sopravvivenza del feto.
Il cromosoma 21
Il cromosoma 21 è il più piccolo di tutto il patrimonio genetico umano e il secondo ad essere stato sequenziato nel Progetto Genoma, l’iniziativa mondiale che ha reso possibile la mappatura completa del DNA.
L’anomalia più diffusa del cromosoma 21 è la trisomia. Ad essa sono associati una sindrome dismorfica, che comporta una serie di caratteristiche fisiche tipiche, e un ritardo psico-motorio costante di grado variabile, oltre ad una serie di complicanze che interessano molti organi e apparati.
La forma lieve della sindrome di Down
Viene definita sindrome di Down a mosaico ed è caratterizzata dalla presenza, nello stesso individuo, di un cocktail di due tipi di cellule, un fenomeno che in biologia è noto come mosaicismo.
Alcune delle cellule contengono i 46 cromosomi fisiologici (con la duplice copia del 21), mentre altre 47 cromosomi (con la presenza di una terza copia del 21).
Quali sono le conseguenze
Le prognosi riguardo l’efficienza intellettiva e le complicanze d’organo dipendono dalla proporzione fra le cellule sane e quelle con la trisomia. Naturalmente, maggiore sarà il numero delle ultime rispetto alle prime, più sfavorevole sarà il quadro clinico.
Si tratta, tuttavia, di un rischio che non può essere calcolato a priori, nemmeno con il supporto della diagnostica sofisticata attualmente disponibile.
Alcune persone con la sindrome di Down a mosaico hanno una sintomatologia molto lieve e un’intelligenza nella norma, mentre per altre le manifestazioni sono praticamente sovrapponibili a quelle delle forma classica.
La presenza di un mosaicismo per la trisomia 21 determina un rischio maggiore rispetto alla norma di avere un secondo figlio affetto dalla sindrome. In questi casi il rischio supera quello corrispondente all’età materna.

Le cause della sindrome di Down
La causa di questa patologia è la presenza del cromosoma 21 in triplice copia, la cosiddetta trisomia 21.
Nel 95% dei casi di pazienti con sindrome di Down sono presenti 47 cromosomi invece dei normali 46.
In una percentuale minima di casi, esiste una triplice copia del 21 ma i cromosomi rimangono 46. Come può succedere?
Quando le cellule si replicano, il loro materiale genetico viene duplicato: una copia rimane nella cellula madre e l’altra finisce nella cellula figlia. Talvolta, nel corso di questi passaggi, si verificano errori. In alcuni casi i cromosomi non riescono a suddividersi correttamente nelle due nuove entità e così si ottengono cromosomi nello stesso numero ma più grandi del normale, un fenomeno denominato non-disgiunzione.
I fattori di rischio
L’età, in particolare quella materna, è il fattore di rischio più importante.
Tutte le altre possibili ipotesi fino ad oggi formulate, che hanno considerato l’esposizione ad agenti chimici, radiazioni ionizzanti, virus, all’effetto di alterazioni metaboliche o endocrine materne, non sono state mai avvalorate da evidenze scientifiche.
Quanto conta l’età materna
Gli studi presenti in letteratura scientifica mostrano come l’incidenza della malattia sia strettamente legata all’età materna al momento del concepimento.
Le ricerche evidenziano come il cromosoma 21 supplementare raramente provenga dal padre.
Pertanto, il rischio di una coppia di concepire un bambino con un cromosoma in eccesso aumenta con l’aumentare dell’età della madre.
L’incidenza globale tra i nati vivi è di circa 1/700, e il rischio di incidenza aumenta gradualmente con l’aumentare dell’età materna.
Sotto i 30 anni il rischio è di un bambino ogni 1.500 nati, a 35-39 è di uno su 280 e a 40-44 è di uno su 70, mentre oltre i 45 è un neonato ogni 38.
Le persone affette da sindrome di Down possono avere figli?
Le donne hanno il 50% delle probabilità di concepire un figlio con la stessa malattia.
Gli uomini sono generalmente sterili.
I sintomi
La sindrome di Down possiede caratteristiche tipiche fisiche e comporta manifestazioni cliniche comuni a tutti i pazienti, anche se non tutte co-presenti.
Le manifestazioni fisiche durante la crescita
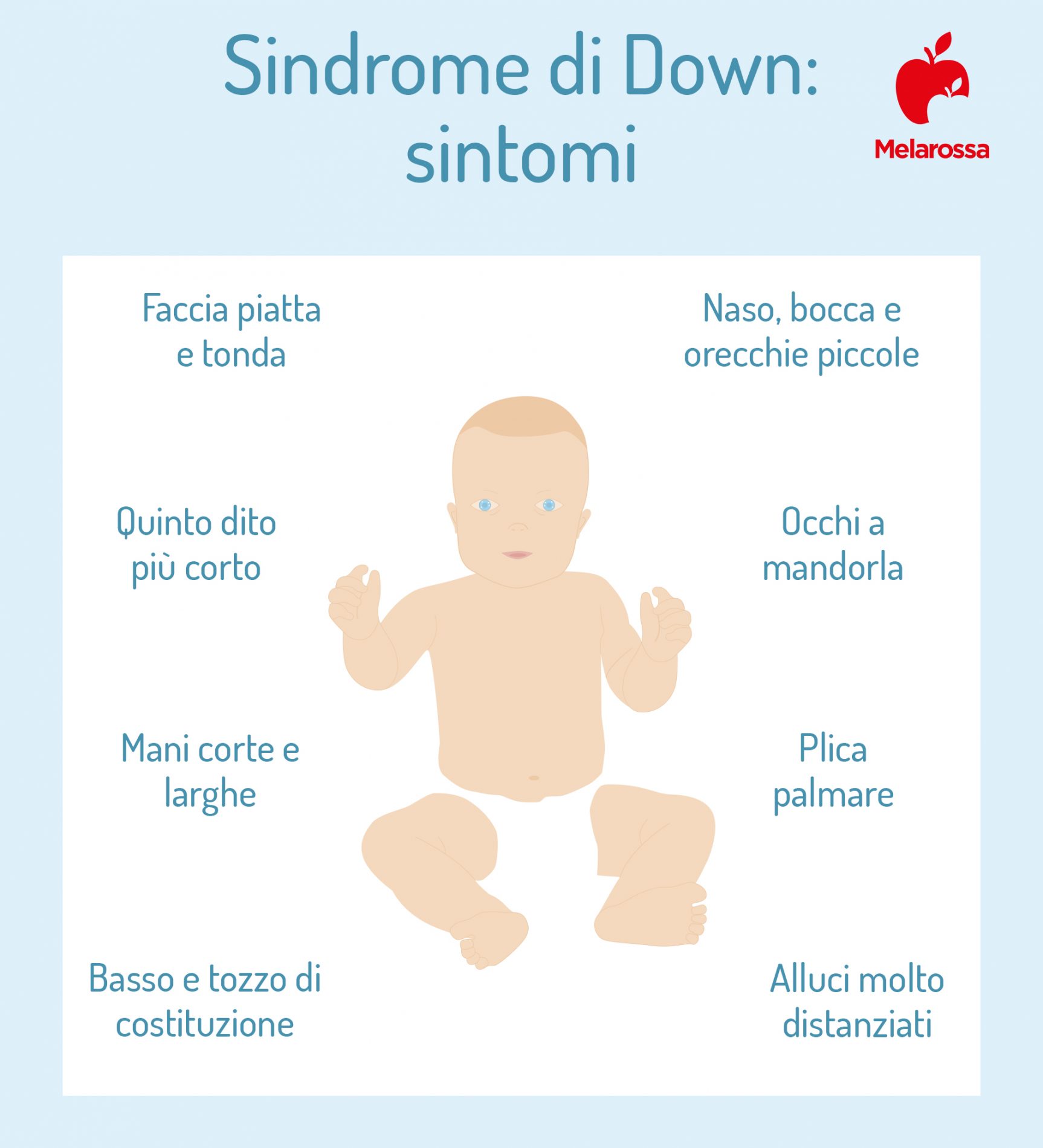
Questa patologia è associata a peculiarità fisiche piuttosto comuni fra le persone che ne sono affette e diffusamente riconoscibili.
La testa è piccola e ha l’occipite (l’osso che costituisce la nuca) piatto. La cute presente in questa area è abbondante e forma una plica accentuata rispetto a quella dei neonati sani.
Gli occhi sono a mandorla, piccoli e obliqui verso l’alto e sono presenti alcune piccole pieghe di pelle al loro angolo interno (una caratteristica definita epicanto). Il bordo palpebrale è rivolto verso l’alto.
Il naso è piccolo e talvolta un po’ schiacciato, a causa del deficit di sviluppo delle ossa nasali. Le orecchie sono minute, a volte leggermente “a sventola” e situate più in basso rispetto alla norma.
La bocca è piccola ma con labbra grosse e corrugate. La lingua protrude attraverso le labbra, perché più grossa del normale. Il palato è alto e stretto e i denti sono soggetti a frequenti malformazioni.
Le mani sono tozze, corte e larghe, e presentano, in corrispondenza del palmo, un solco trasversale unico. Il quinto dito è corto. I piedi hanno dita corte e separate da ampi spazi.
Le manifestazioni cliniche
I bambini con sindrome di Down sono caratterizzati da un ritardo nell’accrescimento e nello sviluppo degli apparati e un ritardo mentale di grado variabile.
Inoltre, hanno caratteristiche riduzioni del tono muscolare e lassità dei legamenti osteo-articolari (ipotonia). Infine, sono soggetti ad una serie di complicanze dovute alle alterazioni presenti nei singoli organi e tessuti, dovute all’anomalia cromosomica.
Le complicanze correlate alla sindrome di Down
Di seguito una panoramica sulle malattie che possono essere presenti nei pazienti.
Complicanze ematologiche
Le persone affette da questa sindrome hanno un rischio aumentato di sviluppare una serie di disturbi del sangue.
Leucemia linfoblastica acuta
Si tratta di un tumore del sangue che interessa le cellule che, sviluppandosi all’interno del midollo osseo, si trasformano in globuli bianchi (linfociti).
Il risultato è la presenza in circolo di una quantità enorme di queste cellule immature (blasti) e una riduzione patologica del numero di linfociti maturi e funzionanti e, dunque, un indebolimento delle difese immunitarie che causa un elevato rischio di infezione.
Lo sviluppo del tumore a livello del midollo osseo ostacola anche la produzione delle altre cellule del sangue, globuli rossi e piastrine. Di conseguenza, i pazienti sviluppano disturbi quali:
- Anemia.
- Comparsa di petecchie.
- Sanguinamenti.
- Emorragie mucosali.
L’accumulo dei blasti nei tessuti periferici è responsabile dell’alterazione nel funzionamento di diversi organi. A livello cerebrale, può provocare:
- Cefalea.
- Vomito.
- Ictus.
- Disturbi della vista, dell’equilibrio, dell’udito e dei muscoli facciali.
Il deposito nel midollo provoca dolore osseo. Nel fegato e nella milza è correlato al rigonfiamento di questi organi (epatosplenomegalia) e dolore addominale.
Lo sviluppo di nuovi farmaci ha permesso un’estensione delle percentuali di guarigione dei pazienti, che nei giovani toccano l’80% e negli adulti il 30-40%.
Leucemia mieloide acuta
È un tumore del midollo osseo a progressione rapida che interessa specificamente le cellule staminali destinate a svilupparsi per dare luogo ai granulociti, elementi del sistema immunitario.
Anomalie cromosomiche come la trisomia 21 e la trisomia 8 aumentano il rischio di sviluppare la leucemia mieloide acuta.
Comporta la comparsa iniziale di sintomi aspecifici, facilmente confondibili con sindromi influenzali o conseguenze di periodi particolarmente stressanti, come:
- stanchezza.
- Perdita dell’appetito.
- Sudorazione notturna.
- Febbre.
La compromissione delle funzioni ematopoietiche del midollo osseo (ossia di produzione delle cellule del sangue, piastrine, globuli bianchi e globuli rossi) causa sanguinamenti frequenti, anemia e un aumento del rischio di infezioni.
Come per la leucemia linfoblastica, si può avere la compromissione degli organi nei quali si depositano le cellule del sangue che non riescono a maturare.
Trombocitopenia
La carenza di piastrine (trombociti) nel sangue è dovuta ad una compromissione della funzione del midollo osseo e causa un rischio di sanguinamento a livello della pelle e delle mucose (in particolare di quella gengivale) e la comparsa di ecchimosi.
Il trattamento del disturbo prevede la prevenzione di tutti i fenomeni che possono aumentare il rischio di sanguinamento, compresa l’assunzione di farmaci che possono avere un effetto sulla coagulazione del sangue.
In condizioni di emergenza, le piastrine mancanti possono essere fornite ai pazienti tramite trasfusioni.
Policitemia neonatale
Si tratta di un disturbo che interessa i neonati e che consiste nella presenza eccessiva di globuli rossi nel sangue. Questa condizione rende il sangue del piccolo più viscoso del normale, ostacolandone il flusso nei capillari.
Spesso il problema viene tenuto sotto controllo semplicemente aumentando la quantità di liquidi ingerita. Quando questa misura non è sufficiente a diluire il sangue, il neonato viene sottoposto ad un prelievo di globuli rossi.
Il piccolo affetto da policitemia ha un colorito più scuro, per via della presenza di una maggiore quantità di emoglobina (la proteina presente nei globuli rossi e deputata al trasporto di ossigeno nel sangue) in circolo. Sono anche soggetti al rischio di convulsioni.
Complicanze a carico del sistema nervoso centrale
Deficit intellettivo
Può comparire con diversa gravità ed è dovuto alla mancanza di un adeguato sviluppo delle funzioni cognitive e adattative della persona. Attualmente, la comunità scientifica tende più a considerarlo come uno stato mentale, piuttosto che un’entità clinica precisa.
Il 60-70% dei casi di ritardo mentale è associato a difetti cromosomici: la sindrome di Down è il più frequente.
Ritardo nelle abilità motorie
Possono essere coinvolte diverse funzioni:
- Motricità (penalizzata anche dall’ipotonia del sistema locomotore).
- Ragionamento.
- Linguaggio.
- Comunicazione.
- Memoria.
- Attenzione.
- Competenze sociali.
I bambini con questa patologia hanno spesso difficoltà nel mantenimento dell’equilibrio, nella coordinazione del movimento, nella strutturazione di sequenze complesse di movimenti, nella comunicazione fra i due emisferi cerebrali. Anche la percezione proveniente dalle strutture nervose periferiche è rallentata.
Ritardo nel linguaggio
Le difficoltà nel linguaggio, che peggiorano nell’età avanzata (quando può subentrare la demenza), possono interferire con il temperamento del soggetto, rendendolo irritabile e collerico.
I deficit nelle capacità linguistiche, che rendono ancora più difficile la comunicazione, sono alla base di molti problemi di inserimento sociale.
Deterioramento cognitivo
L’estensione dell’aspettativa di vita ha fatto emergere una serie di disturbi del sistema nervoso centrale analoghi a quelli che interessano le persone sane nella fase dell’invecchiamento ma che, rispetto a queste, hanno un esordio precoce.
A partire dai 40 anni di età, i soggetti con questa malattia hanno, infatti, un rischio aumentato di problemi neuropsichiatrici, che includono un declino delle facoltà intellettive e le crisi epilettiche, e una marcata riduzione della capacità di elaborare il pensiero.
Alzheimer precoce
La demenza che caratterizza l’invecchiamento delle persone con questa sindrome, in particolare dopo i 50 anni, ha molte analogie con l’Alzheimer.
Dal punto di vista clinico, compare un deterioramento delle funzioni mentali ed emotive, che si manifesta con:
- apatia o inspiegabile eccitamento.
- Irritabilità, collera.
- Regressione nelle abilità acquisite.
- Perdita dell’interesse per la cura e per l’igiene personali.
La demenza, il cui esordio è preceduto generalmente da crisi epilettiche, ha una progressione lenta e graduale. Le prime alterazioni riguardano la memoria a breve termine, il comportamento e l’orientamento spaziotemporale.
Successivamente si manifestano disturbi del linguaggio (afasia), della memoria in generale (amnesia), della percezione e del riconoscimento (agnosi), limitazione nelle attività motorie (aprassia) e nell’esecuzione dei movimenti.
Tutti aspetti che penalizzano significativamente la qualità della vita.
Invece, nelle fasi più avanzate, si può avere un aumento generalizzato del tono muscolare che rende i movimenti impacciati e goffi e la presenza di malattie ricorrenti che, nel tempo, portano il quadro clinico a precipitare.
Le complicanze del sistema digerente
Malformazioni intestinali
Il duodeno è la prima porzione dell’intestino tenue, localizzata subito dopo lo stomaco.
La principale malformazione intestinale presente nei pazienti è l’atresia duodenale, ovvero l’ostruzione completa di questa struttura anatomica. Il 30% dei casi di atresia duodenale riguarda bambini con sindrome di Down.
Questa condizione si realizza già durante la gravidanza. Normalmente, il feto ingerisce liquido amniotico: nel caso sia presente un’ostruzione, tuttavia, il liquido non può scendere nel canale digestivo e si accumula nei tratti alti, dilatandoli. Il risultato è lo scarso sviluppo dell’intestino a valle.
Può essere trattata chirurgicamente subito dopo la nascita.
Malattia di Hirschsprung
Si tratta di una malformazione dei nervi che afferiscono al colon e che determina una penalizzazione più o meno marcata delle sue funzioni, provocando:
- stipsi.
- Gonfiore e dolore addominale.
Il problema è causato da un difetto di migrazione delle cellule nervose specifiche durante la gravidanza. Può essere corretta attraverso un intervento chirurgico, che viene generalmente eseguito dopo la nascita.
Celiachia
La celiachia è una intolleranza nei confronti del glutine causata da un’anomala risposta del sistema immunitario alla sua ingestione.
Questo disturbo ha un’incidenza 10-40 volte superiore nelle persone con la sindrome di Down rispetto alla popolazione generale.
Poiché talvolta può produrre sintomi lievi o addirittura impercettibili, pur esercitando un’azione di infiammazione sulla mucosa intestinale, viene raccomandata ai pazienti l’esecuzione annuale del test specifico.
Le complicanze endocrine
Ipotiroidismo
Le disfunzioni tiroidee sono presenti in una percentuale di pazienti che varia dal 4 al 18%: fra queste, l’ipotiroidismo è la più diffusa.
La causa può essere l’assenza dell’attività tiroidea alla nascita (ipotiroidismo congenito) oppure una reazione autoimmune che aggredisce il tessuto di questa ghiandola determinandone l’indebolimento funzionale (ipotiroidismo autoimmune).
La comunità scientifica non ha chiarito le condizioni precise nelle quali deve essere instaurata la terapia ormonale sostitutiva dell’ormone tiroideo mancante.
Sindrome metabolica, diabete e obesità
A causa della chiara correlazione fra sindrome di Down e le alterazioni del quadro metabolico e lipidico, molte associazioni scientifiche raccomandano specifiche e periodiche valutazioni di pazienti.
I pazienti con questa patologia sono più esposti al rischio di diabete e di obesità, con concentrazione del grasso corporeo soprattutto a livello addominale.
Gli studi effettuati sembrano dimostrare un’interferenza di tipo genetico con i processi metabolici.

Le complicanze oculari
Le persone con questa patologia hanno un rischio aumentato di sviluppare disturbi oculari.
Questi possono essere di tipo motorio (strabismo e nistagmo), di tipo refrattivo, infiammatorio (blefariti), che interessano la morfologia corneale (cheratocono) oppure relativi alla trasparenza dei mezzi diottrici (cataratta).
Cheratocono
Le ricerche dimostrano come il 70% circa dei pazienti abbia una morfologia corneale assimilabile a quella presente nel cheratocono, diagnosticabile come conclamato nel 15% di essi.
Questo suggerisce la necessità di controlli periodici, finalizzati a scongiurare il rischio di compromissione definitiva della capacità visiva.
Cataratta
La cataratta è presente nel 15% dei pazienti dalla nascita (cataratta congenita) e sembra essere dovuta a fenomeni ossidativi che determinano la deposizione di residui che alterano la trasparenza del sistema di lenti naturali dell’occhio.
Le complicanze otologiche
Infezioni auricolari
Il bambino con sindrome di Down, avendo un condotto uditivo più stretto della norma, è più esposto al rischio di sviluppare un’otite media secretiva, ossia un’infezione del condotto con presenza di un versamento di muco non infetto che si deposita dietro la membrana timpanica integra.
L’otite media secretiva può causare una riduzione della capacità uditiva (ipoacusia).
Ipoacusia trasmissiva
È la causa più comune di riduzione dell’udito nelle persone con questa patologia ed è prevalentemente dovuta a otiti medie secretive ricorrenti.
Per evitare il continuo accumulo di liquido dietro il timpano, può essere necessario inserire dei drenaggi di ventilazione, ossia dei tubicini che permettono di drenare il muco impedendone il deposito.
Talvolta l’ipoacusia trasmissiva può essere causata da una malformazione degli ossicini presenti nell’orecchio interno e coinvolti nella trasmissione del suono (otodisplasia): questa forma di riduzione della capacità uditiva può essere risolta con l’applicazione di una protesi acustica a conduzione ossea.
Ipoacusia neurosensoriale
Questo tipo di riduzione dell’udito è meno comune e, quando si verifica, è permanente. La causa è sempre legata ad anomalie dell’orecchio interno, legate al nervo uditivo e alle altre strutture anatomiche che consentono la trasmissione del suono verso il cervello.
Possono essere utili gli apparecchi acustici.
Le complicanze cardiache
Il cuore è composto da 4 spazi, detti camere: 2 atrii (uno destro e uno sinistro) e 2 ventricoli (uno destro e uno sinistro).
Le camere cardiache sono perfettamente separate fra loro, per evitare che il sangue venoso si mescoli con quello arterioso e che si vengano a creare fenomeni di reflusso che causano irregolarità nella gittata cardiaca.
Quando, a causa di malformazioni, i setti non sono integri, si possono avere diverse conseguenze sul piano clinico.
Le complicanze cardiache sono presenti nel 50% dei pazienti.
La sindrome di Down è correlata anche ad alterazioni del setto interventricolare e del setto atrioventricolare.
Difetto del setto atrioventricolare o del setto interventricolare
Nel primo caso a non essere integro è il setto che separa atrii e ventricoli, mentre nel secondo quello che separa i due ventricoli, destro e sinistro.
Le anomalie nel flusso all’interno del cuore che si vengono a creare a causa di questi difetti provocano una congestione del sangue a ritroso, nelle arterie polmonari, che porta a ipertensione polmonare.
Il bambino che nasce con queste malformazioni cresce più lentamente della norma ed è a rischio per infezioni respiratorie ricorrenti e scompenso cardiaco congestizio. Le conseguenze sono tanto più pronunciate quanto maggiore è l’entità del problema.
L’alterazione può ripararsi spontaneamente, se di dimensioni limitate, oppure richiedere l’intervento chirurgico.
Le complicanze osteo-articolari
La maggior parte dei problemi osteo-articolari nei pazienti è dovuta alla lassità dei legamenti articolari e allo scarso tono muscolare (ipotonia).
Il piede piatto
La gravità del problema varia con il grado di ipotonia muscolare e di lassità legamentosa che interessano il paziente.
Il trattamento in genere è conservativo. È possibile utilizzare un plantare semirigido da applicare a calzature normali per stabilizzare il piede ed evitare la comparsa del dolore.
La chirurgia viene raccomandata quando la deformità è tanto grave da compromettere la deambulazione oppure è causa di dolore cronico: in questi casi viene inserito chirurgicamente un supporto nelle ossa del tarso per risollevare l’arco plantare.
La lussazione della rotula
La debolezza del muscolo quadricipite femorale e la lassità dei suoi legamenti causano una instabilità della rotula che tende a spostarsi lateralmente e a causare un’instabilità del ginocchio con tendenza alle cadute.
Lo spostamento della rotula causa uno sfregamento contro il femore e un’usura progressiva della cartilagine interposta, che provoca l’insorgenza di dolore e riduzione dei movimenti.
La fisioterapia può aiutare a irrobustire la muscolatura e favorire il mantenimento in sede dell’osso.
L’instabilità dell’anca
La lussazione frequente dell’anca dovuta ai semplici movimenti del bambino può portare a un danno articolare.
Instabilità della colonna cervicale
L’instabilità della colonna riguarda l’articolazione fra la prima e la seconda vertebra cervicali (articolazione atlanto-assiale) ed è presente nel 15% circa dei bambini con sindrome di Down.
Il rischio è che una scossa o un trauma, anche non gravi, possano danneggiare il midollo spinale, causando lesioni nervose definitive.
Pertanto, ai bambini che praticano sport viene raccomandata attenzione specifica nei confronti di movimenti che possono comportare un pericolo.
Come si diagnostica la sindrome di Down
La malattia può essere diagnosticata esclusivamente attraverso lo studio della mappa cromosomica del soggetto, la cosiddetta analisi citogenetica.
La diagnosi clinica, che ha valore orientativo e indicativo, può invece essere posta sulla base dei tratti fisici tipici, non tutti contemporaneamente presenti, ma che nel loro insieme caratterizzano il fenotipo Down.
Ma è importante sottolineare che queste caratteristiche non sono diagnostiche e possono, singolarmente, essere presenti anche in persone sane.
La diagnosi in gravidanza o dopo la nascita
Il test per la sindrome di Down viene effettuato sul feto attraverso un’analisi citogenetica, che viene eseguita dopo la nascita, su una cultura di linfociti prelevati dal sangue periferico, oppure durante la gravidanza.
Il test praticato in gravidanza viene effettuato su colture di cellule prelevate dai villi coriali o dal liquido amniotico.
Prelievo dei villi coriali
Alcuni villi coriali, strutture parte del tessuto che costituisce la placenta, vengono aspirati all’interno di una siringa e messi in coltura.
L’esame fornisce informazioni riguardo la mappatura genetica e cromosomica del feto e possiede un’accuratezza simile a quella dell’amniocentesi. Tuttavia, il prelievo di villi coriali viene eseguito in epoca più precoce, tra la decima settimana di gestazione e la fine del primo trimestre, permettendo, nel caso in cui sia deciso, un aborto più tempestivo, con una procedura più semplice e meno rischiosa.
Questo esame è ormai una procedura di routine: raramente possono verificarsi errori dovuti alla contaminazione del campione da parte di cellule materne. Il rischio di morte del feto è pari allo 0,2% dei casi, in analogia all’amniocentesi.
Tuttavia, il prelievo dei villi coriali è associato ad un rischio superiore (ma comunque molto basso) di difetti trasversali degli arti e di una condizione definita sindrome oromandibolare-ipogenesia degli arti.
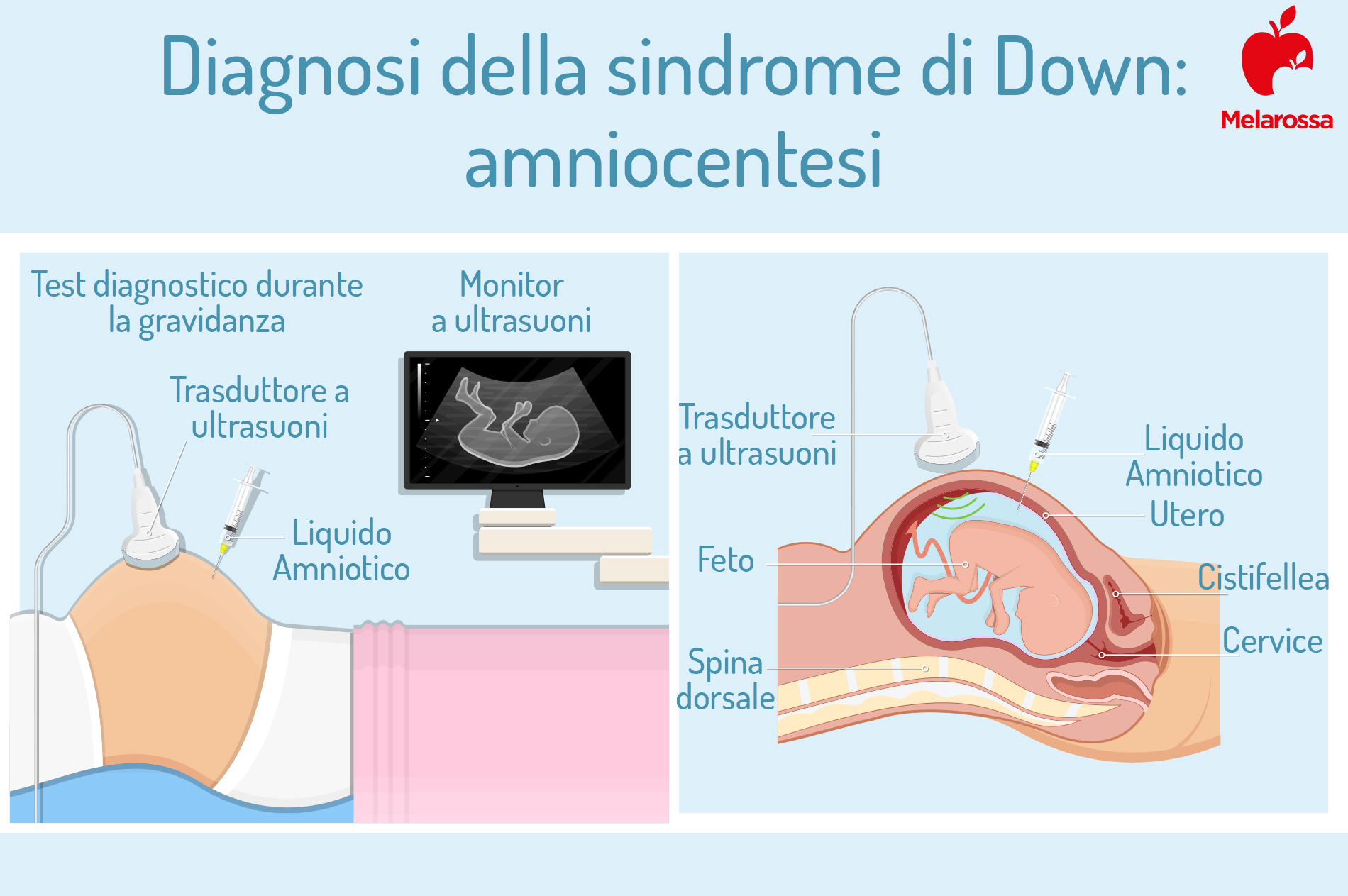
Amniocentesi
Un ago viene inserito attraverso l’addome, sotto guida ecografica, all’interno del sacco amniotico, allo scopo di prelevare il liquido amniotico e le cellule fetali da analizzare.
L’esecuzione è raccomandata dopo la quattordicesima settimana di gestazione, per minimizzare i rischi.
Fra le complicanze materne dell’amniocentesi, l’amnionite sintomatica e condizioni meno preoccupanti come lo spotting vaginale o la perdita di liquido amniotico. In generale, se ci si affida a operatori esperti, il rischio di morte fetale è di circa 0,1-0,2%.
Tritest
È stato osservato che i livelli di alcuni marcatori presenti nel siero materno, nello specifico alfa-fetoproteina, estriolo non coniugato e beta-gonadotropina corionica, si modificano significativamente nel corso del secondo trimestre di gravidanza, se il feto è affetto da trisomia 21.
La combinazione di questi valori, insieme all’età materna e all’epoca gestazionale consente di valutare in modo più personalizzato il rischio di una trisomia. Nel caso in cui la probabilità che sia presente questo disturbo sia significativa, può essere raccomandata l’analisi citogenetica.
Occorre anche precisare che il tri-test non ha valore diagnostico, ma viene utilizzato per orientare la diagnostica.
Ecografia genetica
L’ecografia genetica è un esame ecografico che viene eseguito intorno alla sedicesima-diciottesima settimana per la ricerca dei cosiddetti soft markers per la sindrome di Down, ossia manifestazioni della malattia presenti singolarmente anche in feti sani (in questo caso privi di significato patologico) ma che possono rappresentare un criterio orientativo per la diagnosi.
Quando viene evidenziato un soft marker viene valutata dettagliatamente l’anatomia fetale, per evidenziarne altri, eventualmente, o altre anomalie associate. Se sono presenti due o più marker in associazione viene raccomandata l’amniocentesi, uno degli esami a valore diagnostico per la malattia.
Focus iperecogeno
La presenza di un’area evidenziata dall’ecografia (anche definita golf ball) localizzata all’interno del ventricolo sinistro è uno dei soft markers. È dovuta alla calcificazione di uno dei muscoli papillari del cuore, che hanno la funzione di tenere ancorati i lembi della valvola atrio-ventricolare.
Dilatazione renale pelvica
La dilatazione dei bacinetti renali (le strutture di raccordo fra i reni e gli ureteri) è nota anche come pielectasia e, se superiore ai 5 millimetri, pone l’indicazione per una valutazione dettagliata dell’anatomia fetale, tramite l’esecuzione di un’ecografia di secondo livello.
Intestino iperecogeno
L’intestino appare più bianco all’osservazione ecografica: si tratta di un marker presente con frequenza tripla nei feti con sindrome di Down.
Cisti dei plessi corioidei
All’ecografia, si osserva la presenza di aree anecogene (ovvero più scure) all’interno dei plessi corioidei, strutture che si trovano all’interno dei ventricoli cerebrali con la funzione di produrre il liquor cefalorachidiano.
Altri soft markers
Oltre a quelli sopraindicati, possono essere presenti segni ecografici come il femore corto, la plica nucale ispessita, il deficit di sviluppo delle ossa nasali e l’arteria ombelicale unica.
Prognosi
La prognosi dipende dalle caratteristiche individuali del paziente e dal grado di compromissione dei diversi apparati.
Per la sindrome non è disponibile alcun trattamento farmacologico, ma devono essere curate le complicanze che possono comparire e riguardare diversi sistemi.
Riabilitazione
La riabilitazione garantisce al bambino affetto dalla malattia uno sviluppo armonico e un buon inserimento scolastico, sociale e lavorativo.
Perché i risultati siano ottimali deve essere intrapresa fin dai primi mesi di vita ed eseguita appieno nei primi 3 anni, fondamentali per la realizzazione delle abilità cognitive e di socializzazione.
Se è vero, da un lato, che la crescita e lo sviluppo del bambino Down si verificano in ritardo rispetto alle consuete tappe del bambino sano, è anche vero che gli step sono uguali. I bambini affetti dalla sindrome imparano a camminare, parlare, correre e giocare.
Rimane invece il problema del deficit cognitivo, che deve essere affrontato con un intervento educativo globale e personalizzato che favorisca un’interazione dinamica tra le sue potenzialità e l’ambiente circostante.
Scelta dell’aborto
Il prolungamento significativo dell’aspettativa di vita delle persone con sindrome di Down sposta l’attenzione sull’opportunità della scelta dell’aborto.
In Europa, Islanda e Danimarca sono fra i Paesi nei quali la percentuale più elevata di donne diagnosticate con trisomia 21 sceglie l’interruzione della gravidanza. Negli ospedali di Reykjavik nascono al massimo uno o due bambini all’anno con questo difetto cromosomico.
Il Belgio è stato il primo Stato a farsi carico del costo dell’analisi citogenetica.
In Italia, la normativa a riguardo offre la possibilità di ricorrere all’aborto fino al termine della gestazione in caso di sindrome di Down diagnosticata con esami specifici. Ogni anno nel nostro Paese vengono alla luce 1.200 piccoli con questa malattia e le stime dicono che l’aborto viene scelto nel 90% circa delle diagnosi.

Sindrome di Down e qualità di vita
Via via che si procedeva con il miglioramento del controllo clinico delle manifestazioni della malattia, si è fatto strada il concetto di qualità di vita nella realtà quotidiana dei pazienti, promosso da diversi aspetti sanitari e sociali.
In primo luogo, il principio di normalizzazione: senza negare le inevitabili differenze in termini di percezione e tempistiche, ai bambini con la sindrome di Down, la società ha offerto le medesime opportunità presentate a tutti gli altri. Questo ha avuto come conseguenza un aumento del grado di inserimento dei pazienti nella società.
In seconda battuta, sono stati effettuati enormi progressi nella conoscenza scientifica sulla struttura del cromosoma 21 in esubero e sulle conseguenze specifiche legate alla sua presenza.
Oggi le persone con questo problema hanno mediamente cura della propria salute generale, aderiscono ai programmi elaborati per massimizzare lo sviluppo delle loro abilità, frequentano scuole inclusive e affrontano più o meno tutti gli step della vita dei coetanei.
La sfida non è terminata, ma l’obiettivo adesso è il raggiungimento della massima autonomia personale.
L’aspettativa di vita
L’aspettativa di vita di un soggetto con sindrome di Down nato nel 1929 era di 9 anni, nel 1947 di 12-15 anni, nel 1961 di 18,3 anni. n Italia, come in Europa, oggi l’aspettativa di vita è pari a 62 anni: l’80% delle persone raggiunge i 55 anni e il 10% arriva ai 70.
Il fatto che i pazienti possano oggi invecchiare fa emergere tutte le patologie legate alle fasi più tardive della vita, in particolare le demenze.
L’estensione della sopravvivenza viene attribuita alla tempestività nella cura delle complicanze associate alla malattia e ai programmi di riabilitazione e inserimento sociale. Basti pensare che la presenza di malformazioni cardiache può addirittura quintuplicare il rischio di morte del paziente.

Sindrome di Down: cenni storici
Malgrado sia caratterizzata da aspetti molto tipici e riconoscibili, questa sindrome è stata identificata come entità specifica solo poco più di un secolo fa.
Venne descritta per la prima volta nel 1838 dallo psichiatra francese Jean Etienne Esquirol, che ne individuò le caratteristiche somatiche salienti: fessura palpebrale obliqua, radice del naso depressa, testa piccola e statura bassa.
In realtà sono state rilevate prove della malattia nei resti di persone vissute circa 1.500 anni fa.
Il mongolismo
Un’acquisizione destinata a lasciare il segno nella visione sociale della malattia è stata raggiunta dall’antropologo tedesco J.F. Blumenbach nel 1776.
Nel suo scritto “De generis umani variegate nativa liber” egli descrisse le diverse razze umane. In questo ambito, individuò nella razza mongola una serie di caratteristiche presenti anche nelle persone affette da alcuni tipi di ritardo mentale.
Descrisse:
- Guance rotonde ed estese ai lati.
- Fessure oculari sottili, oblique e allungate verso l’alto.
- Occhi più distanziati rispetto alla norma.
- Labbra grandi e corrugate.
- Naso piccolo.
- Pelle poco elastica.
Dalle sue osservazioni nacque il concetto di “mongolismo”.
Blumenbach aveva anche notato che le persone affette da questo genere di disturbo non lo sviluppavano durante la crescita, ma nascevano con esso.
Le cause
Negli anni successivi il medico britannico John Langdon Down si occupò attivamente dello studio della sindrome, che proprio da lui prese il nome, attivando anche diversi programmi sociali per i pazienti.
La comunità scientifica si occupò delle possibili cause. Inizialmente gli studiosi osservarono che spesso i bambini che nascevano con questo problema avevano un genitore malato di tubercolosi. Quindi pensarono ad un possibile ruolo dell’infezione nella genesi della sindrome.
In seguito, si notò anche che i pazienti erano spesso gli ultimi figli di famiglie numerose. Questo alimentò il sospetto che l’età dei genitori potesse essere fra i fattori di rischio per la malattia.
Negli anni ’30 del secolo scorso si giunse a determinare che questo tipo di sindrome era provocata da un’alterazione cromosomica. Ma fu solo nel 1959 che si scoprì che il problema era la presenza di un cromosoma supplementare.
A seguito delle informazioni emerse nel corso delle ricerche, nel 1961 un gruppo di scienziati di fama internazionale pubblicò un articolo sulla rivista scientifica The Lancet, nella quale chiedeva di abolire il termine “mongolismo”.
La società rispetto alla sindrome di Down
Storicamente le persone con questo difetto cromosomico sono state vittime di isolamento sociale, che ha reso ancora più complicato il loro sviluppo e acuito il ritardo mentale.
Nel secolo scorso, il miglioramento delle conoscenze mediche e la progressiva individuazione di trattamenti per le complicanze di questa patologia hanno reso possibile un aumento della qualità di vita dei pazienti e un’estensione della sua aspettativa.
Di pari passo con le acquisizioni scientifiche, l’inclusione sociale delle persone con sindrome di Down è aumentata, favorendone un progressivo inserimento nel tessuto sociale e stimolandone lo sviluppo fisico e intellettivo.
Oggi la sfida principale è quella della massimizzazione dell’autonomia personale dei pazienti.
Fonti
- Invecchiamento precoce nella sindrome di Down: il ruolo dell’educatore – I. Gaio – Università degli Studi di Padova.
- Mongolism – G. Allen et al – The Lancet, 1961.
- Aspettative di vita nelle persone con sindrome di Down – National Institute for Public Policy Analysis, 2014.
- Tomography and topography findings in patients with Down Syndrome – P. Binder – JAMA, 2018.